Why molecular discovery needs a better generative engine
A familiar pattern in early discovery is that teams can search huge chemical libraries, yet still spend weeks cycling through “close variants” because the practical search space is much larger than what can be enumerated, purchased, or manually designed. Traditional workflows lean on heuristics, fragment rules, and human intuition to propose compounds, then rely on docking and assays to reject most of them. The bottleneck is not only screening capacity; it is the ability to propose candidates that are novel, relevant to the target, and plausible to make.
Generative models promised to widen that funnel, but many approaches behave like idea generators that repeat training-set motifs or optimize the easiest-to-game proxy scores. When a model outputs invalid structures, unstable tautomers, or molecules that satisfy a property predictor while ignoring chemistry, the cost shifts downstream into triage and synthesis planning. A better generative engine has to produce chemically coherent candidates, explore beyond familiar series, and stay steerable under real constraints like ADMET risk, IP space, and synthetic accessibility.
Diffusion models are attracting attention because they can be trained to learn a smooth, controllable notion of “how molecules vary,” then sample from it in a way that is less prone to the brittle shortcuts that show up in some older generators. That does not remove the need for docking, assay confirmation, or retrosynthesis checks, but it can change the starting point: fewer random shots, more targeted proposals, and a clearer interface for conditioning on what the team actually needs. The practical question is whether that shift reduces iteration time without inflating validation work.
What makes diffusion models different from VAEs and GANs
When a VAE proposes a molecule, it usually compresses chemistry into a fixed latent “summary,” then decodes in one shot. That’s fast, but the model can learn a blurry latent space where small moves do not reliably map to sensible structural edits, and it may drift toward average-looking chemistry. GANs can make sharper samples, but training often becomes a stability problem: the generator learns a narrow slice of what looks valid, and diversity collapses—especially when you add strong objectives.
Diffusion flips the game. It learns how to iteratively denoise a corrupted structure, so sampling becomes a sequence of small, correctable steps. That tends to make exploration smoother and conditioning easier, because the model can “steer” each step rather than gamble on a single decode.
The trade-off is compute and engineering: iterative sampling is slower than one-pass generators, and you still need careful validation to avoid plausible-looking but wrong chemistry.
Choosing how a molecule is represented changes everything
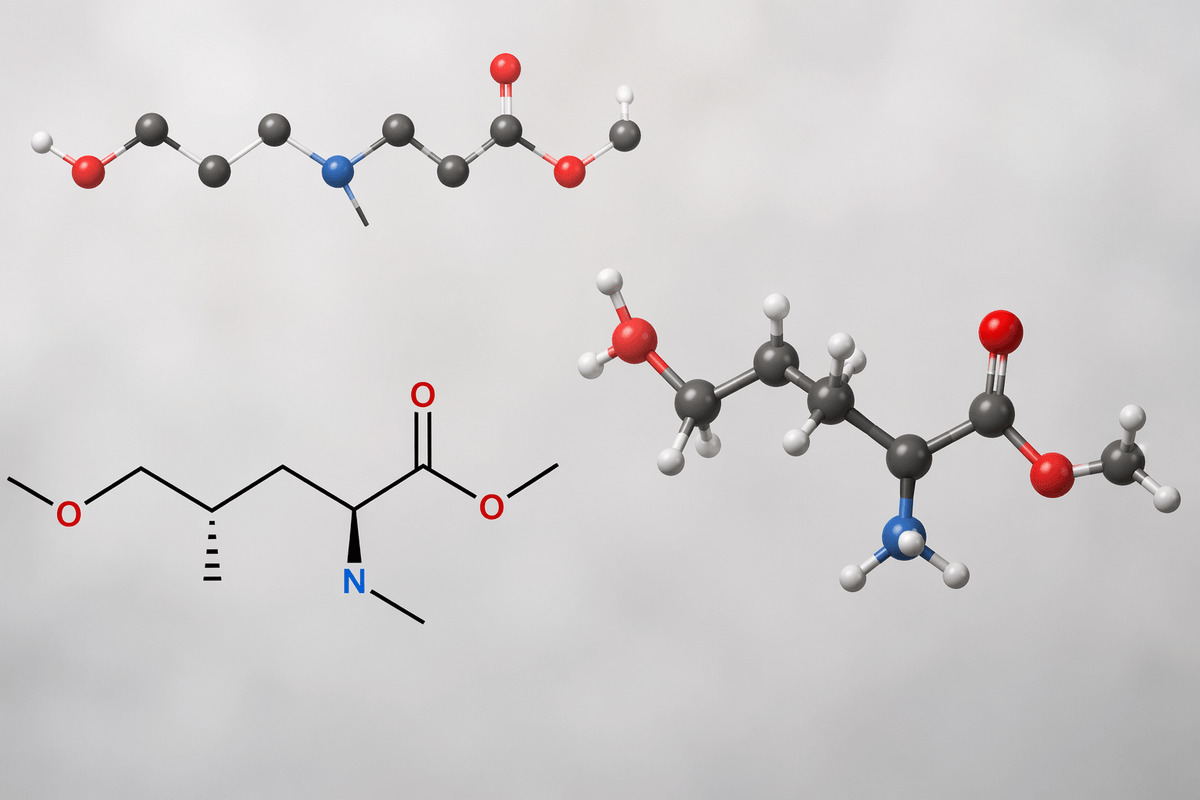
A familiar snag appears the moment you ask “what exactly is the model generating?” If it is SMILES, tiny syntax mistakes create invalid molecules, and even valid strings can hide chemistry issues like awkward valence patterns that only show up after sanitization. If it is a 2D graph, you avoid string grammar but still have to ensure bond orders, charges, and stereochemistry remain consistent, especially under small iterative edits.
3D representations feel closer to what docking and physics care about, but they introduce their own friction: conformers are not unique, coordinates must respect rotational/translation invariance, and you still need to decide whether you are generating a ligand alone or a protein–ligand complex. In practice, the “best” representation is often the one that matches your validation loop and constraints, because every mismatch becomes downstream triage time and compute cost.
How conditioning turns generation into property-driven design
A medicinal chemist rarely wants “a new molecule” in the abstract. They want a molecule that keeps a hinge-binding motif, shifts a pKa window, reduces clearance risk, avoids a liability substructure, or fits a binding site without blowing up lipophilicity. Conditioning is the mechanism that turns diffusion from open-ended sampling into a controllable proposal engine: each denoising step is guided by signals like desired properties, similarity to a starting scaffold, or constraints tied to a target pocket.
In practice, that guidance can come from labels in the training data (activity bins, measured solubility), from learned predictors (e.g., logD, TPSA, hERG risk), or from structure-based objectives when generating in 3D. The model is not “optimizing” in one leap; it is being nudged repeatedly, which makes it easier to trade off competing requirements—potency versus permeability, novelty versus series continuity.
If the property model is biased, poorly calibrated outside its domain, or easy to exploit, diffusion will still find loopholes. Strong constraints also narrow diversity, and the best-looking outputs can be the ones most likely to fail synthesis planning or real assays.
The metrics that matter before you trust the outputs
You can usually tell when a generator is being judged on the wrong scoreboard: it produces lots of “valid” molecules, but your chemists still reject most of them on first glance. Start with basic validity and uniqueness, then move quickly to what affects decisions: novelty versus your internal and public reference sets, scaffold diversity (not just analog count), and property distributions compared to the project’s acceptable windows. A model that matches training-set property histograms but never leaves known chemotypes is not expanding your search.
Track synthesizability with a retrosynthesis or route-feasibility pass rate, not a single heuristic score, and record how often docking or pharmacophore filters agree with the intended conditioning. Most importantly, evaluate hit rate under your actual triage stack—sanitization, filters, docking, and expert review—because iterative sampling can inflate compute cost, and the “pretty” molecules are often the most expensive to falsify.
Where diffusion actually saves time in the discovery pipeline
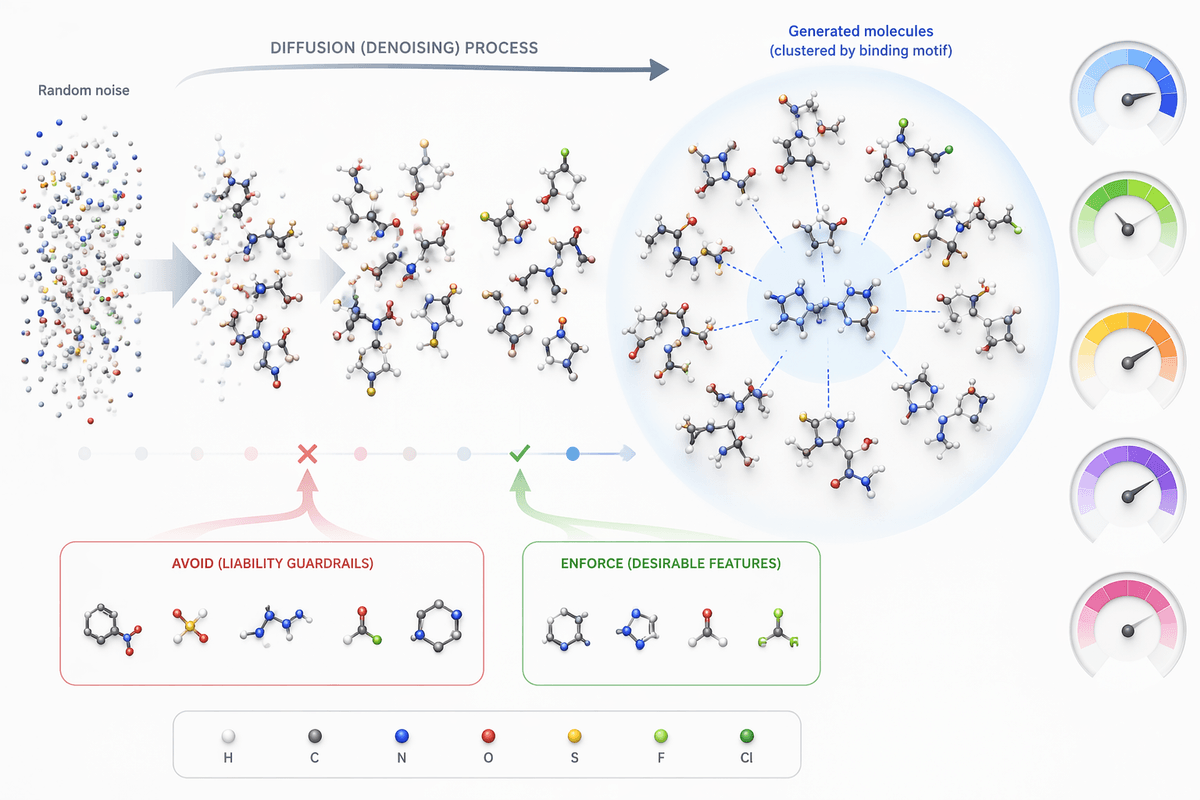
The time savings show up when you are stuck in the “proposal bottleneck”: too many plausible ideas, too few that survive your triage stack. Diffusion helps most when it can generate batches that already respect your project’s guardrails—stay near a known binding motif, avoid specific liabilities, and sit inside rough property windows—so fewer molecules die in sanitization, rule filters, or first-pass docking. That matters in hit expansion, scaffold hopping within a target class, and rapid SAR branching when the team wants diverse, testable hypotheses rather than another handful of close analogs.
It does not usually replace the slow steps. You still pay for docking, free-energy work if you use it, retrosynthesis planning, and real synthesis and assays. Iterative sampling also adds compute and engineering overhead, so the practical win is reducing human review and downstream “obvious rejects,” not eliminating validation.
Common failure modes: invalid chemistry, collapse, and wishful constraints
A team will often see “high validity” in reports, then discover the molecules fail basic sanitization once you standardize charges, aromatization, and stereochemistry. Diffusion can still leak invalid valences, unstable tautomers, or strained rings—especially when the representation is SMILES-like or when you push guidance hard. The practical cost is not academic: every invalid or ambiguous structure becomes extra RDKit cleanup, manual review, and wasted docking and synthesis-planning cycles.
Mode collapse shows up differently than in GANs, but the symptom is familiar: many samples, few distinct ideas. Strong conditioning, narrow training data, or a heavy similarity constraint can trap sampling around a small set of chemotypes, giving the illusion of productivity while your scaffold diversity barely moves.
Then there are wishful constraints: a property predictor says “good,” but the molecules exploit model blind spots, violate developability in ways you did not encode (reactivity, metabolic soft spots), or look synthesizable until retrosynthesis fails. Tighten constraints with real triage signals, not just better prompts.
A pragmatic starting plan for teams adopting diffusion
A pragmatic start is a narrow pilot tied to a real decision: generate hit-expansion ideas around one series, or propose scaffold hops for a well-characterized pocket, with a fixed triage stack (sanitization, filters, docking, retrosynthesis, and a chemist review rubric). Choose a representation that matches your downstream checks, and treat conditioning as “soft steering,” not a promise—use property models you already trust and measure how often they agree with later docking or assays.
Budget for practical friction: iterative sampling compute, integration work, and data cleanup will dominate week one. Set success criteria in pipeline terms (route-feasible rate, scaffold diversity after triage, and how many proposals a chemist would actually put on a synthesis list), then stop or scale based on that evidence.